
Introduction — a lab morning, a stack of results, and a question
I still remember a humid July morning in 2019 when I walked into a small clinic lab and a tray of test strips steamed like warm bread under a hot lamp; the scent of ethanol and heated plastic was sharp and oddly comforting. In that moment I was thinking about biological evaluation — how raw data, tissue staining, and a single in vitro assay can decide whether a device proceeds or stalls. The scenario was simple: three assays, two conflicting signals, and a sponsor who had spent six months and $120,000 on tests that regulators flagged as inconclusive (it stung). What went wrong, and how do we write reports that actually tell the right story to reviewers and engineers? I want to take you through practical fixes, based on hands-on work over the last 18 years in device testing and regulatory submissions, so you can avoid repeated delays and rework. Read on for clear steps and concrete examples that I use when preparing a biological evaluation report — and yes, I’ll include the parts people often skip. –>
Digging deeper: where conventional reports fail
I want to start with the document we all wrestle with: the biological evaluation report. From my experience, the core flaws are not missing data but weak linkage between material characterization, test method, and clinical risk. Too many reports present cytotoxicity, sensitization, and irritation results as separate chapters rather than as a risk chain tied to the device’s intended use. I audited a premarket submission for a polymer-coated catheter in Boston in March 2018 where ISO 10993 tests were performed, but extraction conditions did not reflect the clinical contact time; the result: reviewers questioned relevance and put the file on hold for nine months. That delay cost the sponsor real market time and roughly $85,000 in repeat testing—no one enjoyed that. Technical misalignment like this is common when teams separate materials science from biological endpoints.
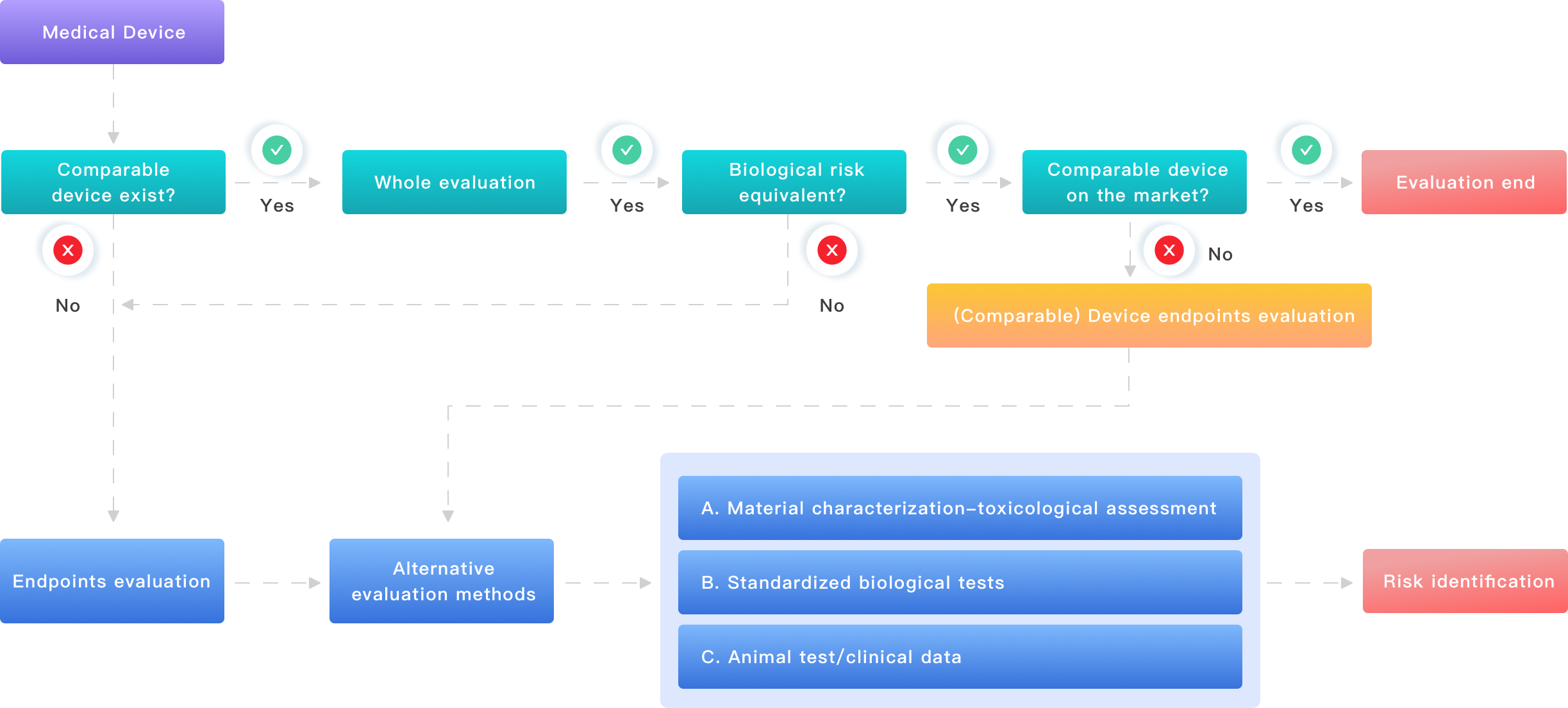
Another frequent problem is inconsistent sample handling. I have seen sterilization validation done one month before material lots were finalized, then hemocompatibility run on a different lot — the variance showed up as a 12% change in clotting markers. The lab reported clean results, but the device vendor later faced a query about lot-to-lot variability. These errors are avoidable with a simple checklist and a culture that ties each biological assay back to a defined worst-case material extraction and clinical exposure scenario. Use terms like extraction vehicle, contact duration, and positive/negative controls explicitly and consistently. Trust me, I’ve been there — small omissions compound quickly and become big regulatory questions.

Why do reports miss the mark?
Often because teams treat reports as a record rather than a narrative. A good biological evaluation report explains why each test matters to patient safety and device performance. It maps materials, sterilization, and clinical use into a clear risk matrix and then shows how the data reduce that risk. When that mapping is absent, reviewers infer uncertainty — and uncertainty invites more testing.
Forward-looking: principles and metrics for better reporting
Looking ahead, I favor a principles-first approach to biological evaluation. Start with clear worst-case definitions for contact (time, temperature, surface area), then design tests around those boundaries. New tools—like targeted extractables profiling and advanced in vitro cytokine panels—help us close evidence gaps faster. I advised a client in 2021 on a cardiovascular scaffold where targeted extractables identified a trace stabilizer that standard cytotoxicity missed; addressing that single compound reduced the need for two animal studies and shortened review time. The principle is simple: use hypothesis-driven testing to avoid shotgun campaigns of assays that add cost but not clarity. Apply ISO-guided selection, but tailor methods to the product’s contact profile.
For practical application, prioritize early integration of material scientists, toxicologists, and regulatory specialists during design freeze. I recommend running a focused extractables study (GC-MS/LC-MS) before full biocompatibility work. That study often highlights compounds that change with sterilization (gamma vs. EtO) and points to appropriate extraction conditions for cytotoxicity and sensitization assays. In short: anticipate changes, test them, and document the rationale. — and yes, this changes timelines but usually reduces total time to clearance.
What’s Next: three metrics to guide decisions
When you pick a path forward, measure impact. I suggest three evaluation metrics to decide on next steps: 1) Relevance ratio — the percent of assays that directly map to a defined clinical exposure scenario; 2) Variability index — measured lot-to-lot differences in key markers (aim for <10% where possible); 3) Time-to-resolution — days from reviewer query to submission of clarifying data. Track these for each project. They give you hard numbers to discuss with leadership and with labs.
Closing guidance from 18 years in the room
I have over 18 years working on device biology, from bench assays in Cleveland to dossier reviews in Brussels. I prefer direct fixes: define worst-case exposure, lock material lots before biocompatibility testing, and run targeted extractables early. Specific details that helped me: a 2017 case where switching sterilization from gamma to EtO reduced polymer embrittlement by 14% and avoided a costly redesign; a 2020 review where a clear table linking assay endpoints to clinical risk cut reviewer questions by half. These are not abstract claims. They are outcomes I have seen and measured.
To close practically, here are three metrics you can apply right away: relevance ratio, variability index, and time-to-resolution. Monitor them across projects and you will spot patterns before they become problems. I stand by these steps because they work in real-world submissions and they save time and money. If you want an example protocol or a checklist from a recent case, I can share it — I keep templates from a November 2019 submission that met a CE review in under 60 days. For technical support or device testing services, consider partners who understand both the assays and the regulatory story, such as Wuxi AppTec Medical device testing.